|
 Oxazolidinone
Oxazolidinone
Antibiotics New classes of antibacterial agents with novel mechanisms
of action are urgently needed to combat the increase in multidrug resistant
infections. Recent reports indicate that in 1998, at least 21% of all
nosocomial enterococcal infections in US hospitals were due to vancomycin-resistant
enterococci (VRE). The oxazolidinones, a new class of totally synthetic
antibacterial agents, are activity against a variety of clinically impotent
susceptible and resistant Gram-positive organisms such as methicillin-resistant
Staphylococcus aureus (MRSA), VRE, and penicillin-resistant Streptococcus
pneumonia (PRSP). Scientists at DuPont originally discovered this class
of agents in the late 1980's. However, development of DuP-721, the drug
candidate that emerged from these initial studies, was discontinued following
Phase I clinical trials. Subsequently, researchers at Pharmacia and Upjohn
identified two clinical candidates, eperezolid and linezolid. Linezolid
is currently marketed for the treatment of multidrug resistant Gram-positive
infections such as nosocomial and community-acquired pneumonia and skin
infections.
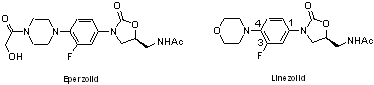
Several SAR studies of the oxazolidinones have demonstrated a high tolerance
for structural variation at the 4-position of the phenyl ring, while the
oxazolidinone ring is essential for its activity. Based on these reports,
we are planning to develop a new oxazolidinone antibacterial agent modifying
the morpholine moiety of linezolid with various functional heterocycle
rings.
(Written
by Dr. Young-Hwan Ha / Senior Researcher)
PDF Inhibitors
Introduction : The emergence of bacterial pathogens that are resistant
to multiple classes of existing antibiotics has created an urgent demand
for new antibacterial agents with novel mechanisms of action. In bacteria,
protein synthesis is initiated by formyl-methionyl-tRNA. As a result,
all nascent polypeptides carry transiently a formylated N-terminus. Peptide
deformylase (PDF) catalyzes the removal of the formyl group from those
polypeptides, and subsequently methionine aminopeptidase hydrolyzes N-blocked
polypeptides to produce mature proteins. Unlike the bacterial protein
synthesis, cytosolic protein synthesis in mammalian cells does not involve
the formyl group and PDF is apparently absent. Thus, the essential role
of PDF in all bacterial protein synthesis provides a rational basis for
selectivity, making it an attractive broad-spectrum antibiotic target.
Current Status : We have constructed non-peptidic benzoic acid-based
small focused library as PDF inhibitors and evaluated its PDF inhibitory
activity by PDF-formate dehydrogenase coupled assay. High throughput screening
has been also carried out to find hits with novel scaffolds. Among benzoic
acid-based focused library, a few hits are as potent as actinonin, a potent
natural PDF inhibitor, in PDF inhibitory activity. We have gained some
insight into the structure activity relationship of non-peptidic benzoic
acid-based inhibitors. These results will be helpful in designing new
non-peptidic PDF inhibitors.
(Written
by Dr. Jong-Kook Lee / Senior Researcher)
|


